Statistical methods for functional contextualization of genetic associations
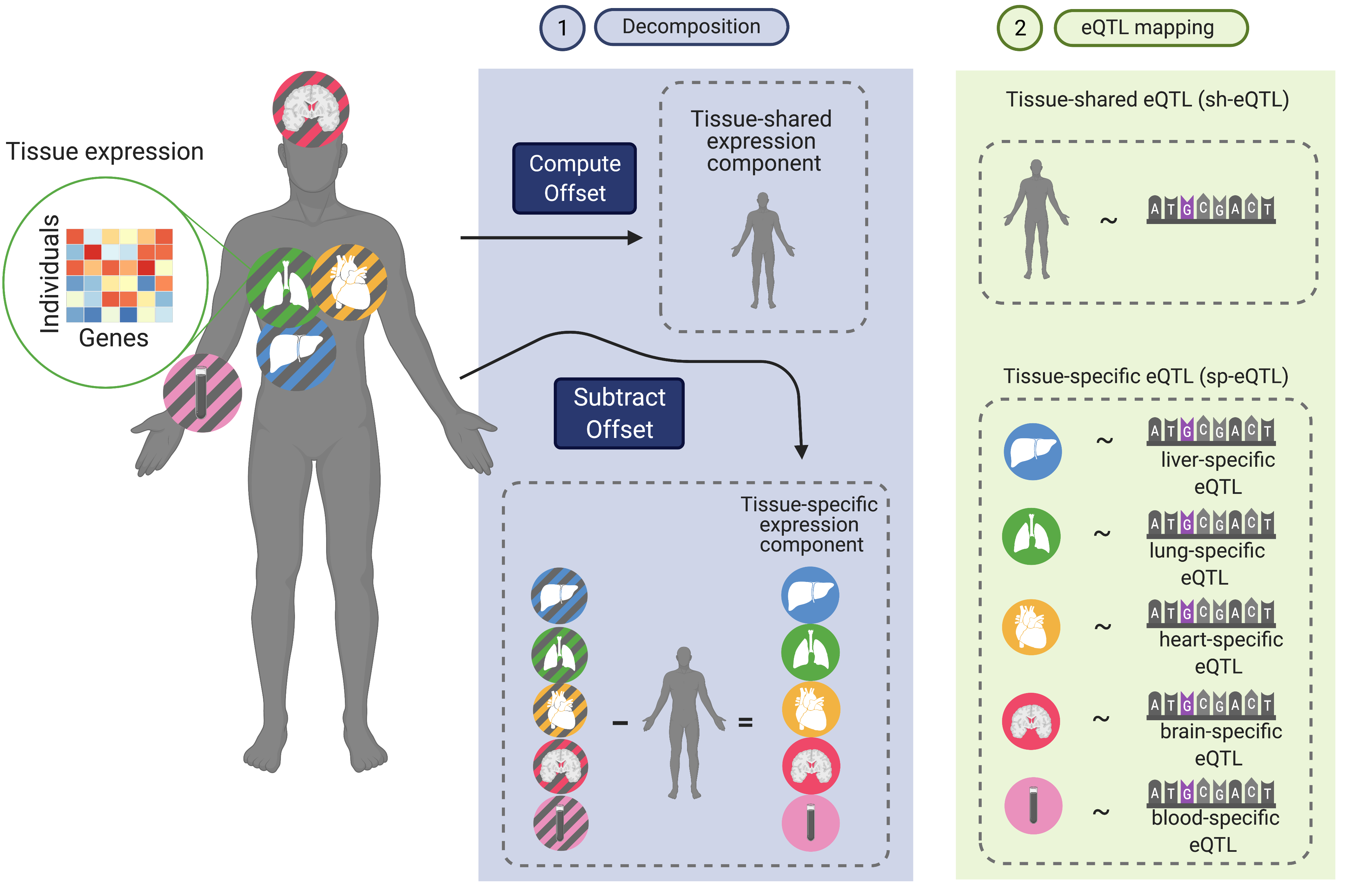
The vast majority of the loci identified from genetic association studies are located outside coding regions, complicating their mechanistic understanding. Our lab develops statistical methods and computational tools for contextualizing genetic associations by leveraging large-scale multi-context functional genomic data (e.g., Genotype Tissue Expression Project, Human Cell Atlas) have emerged as an attempt to find the right genes and contexts for disease-associated variants. Specifically, we have recently developed FastGxC, a computationally efficient and statistically powerful method for detecting context-specific QTL effects in multi-context genomic studies with shared noise. We have applied FastGxC to bulk multi-tissue and single-cell RNA-seq studies to produce the most comprehensive tissue- and cell-type-specific eQTL maps to date. Together with collaborators at UCLA, we have also developed CONTENT, an extension of FastGxC to cross-context transcriptome-wide association studies to increase power to identify genes associated with complex traits and their tissue and cell types of action.